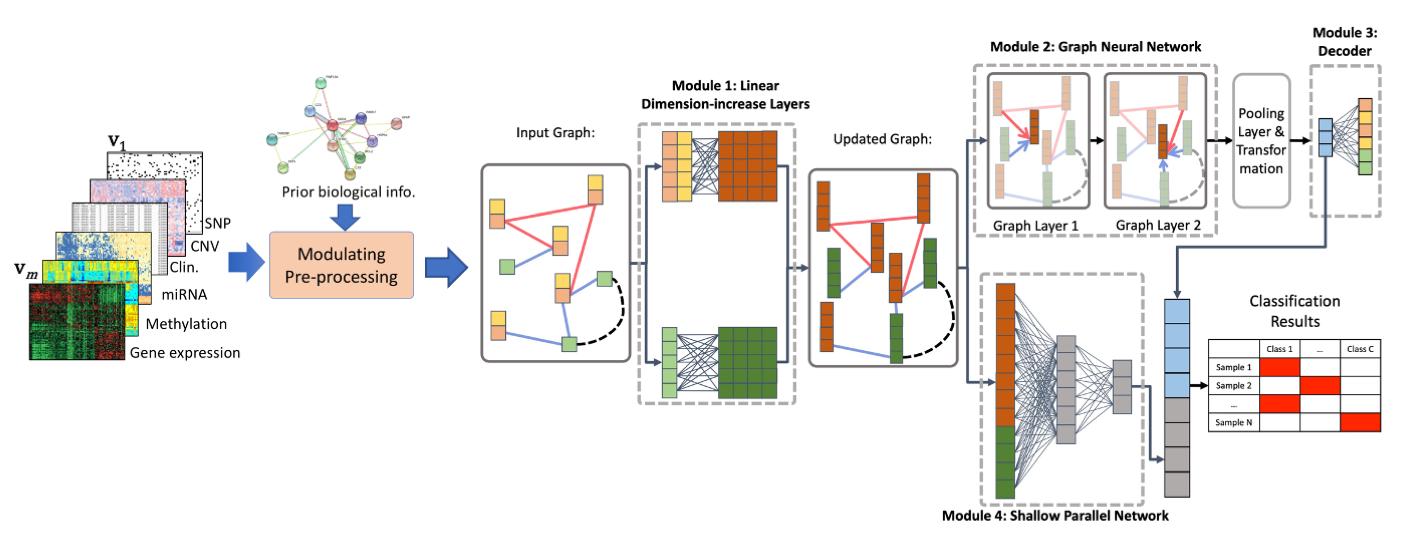
Integrating genomic data to identifying candidate biomarkers and building phenotypic predictive models for cancer studies.
With advances in technologies, huge amounts of multiple types of high-throughput genomics data are available. Large consortiums, such as The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC), have generated several high throughput genomics data types for hundreds of sample on tens of cancer types, which are publicly available.These data have tremendous potential to identify new and clinically valuable biomarkers to guide the diagnosis, assessment of prognosis, and treatment of complex diseases, such as cancer. Integrating, analyzing, and interpreting big and noisy genomics data to obtain biologically meaningful results, however, remains highly challenging. Developing new computational methods based on statistical machine learning and integrative analysis of genomics data is required to facilitate the identification of a short list of biologically meaningful candidate drivers of anti-cancer drug resistance from an enormous amount of heterogeneous data.
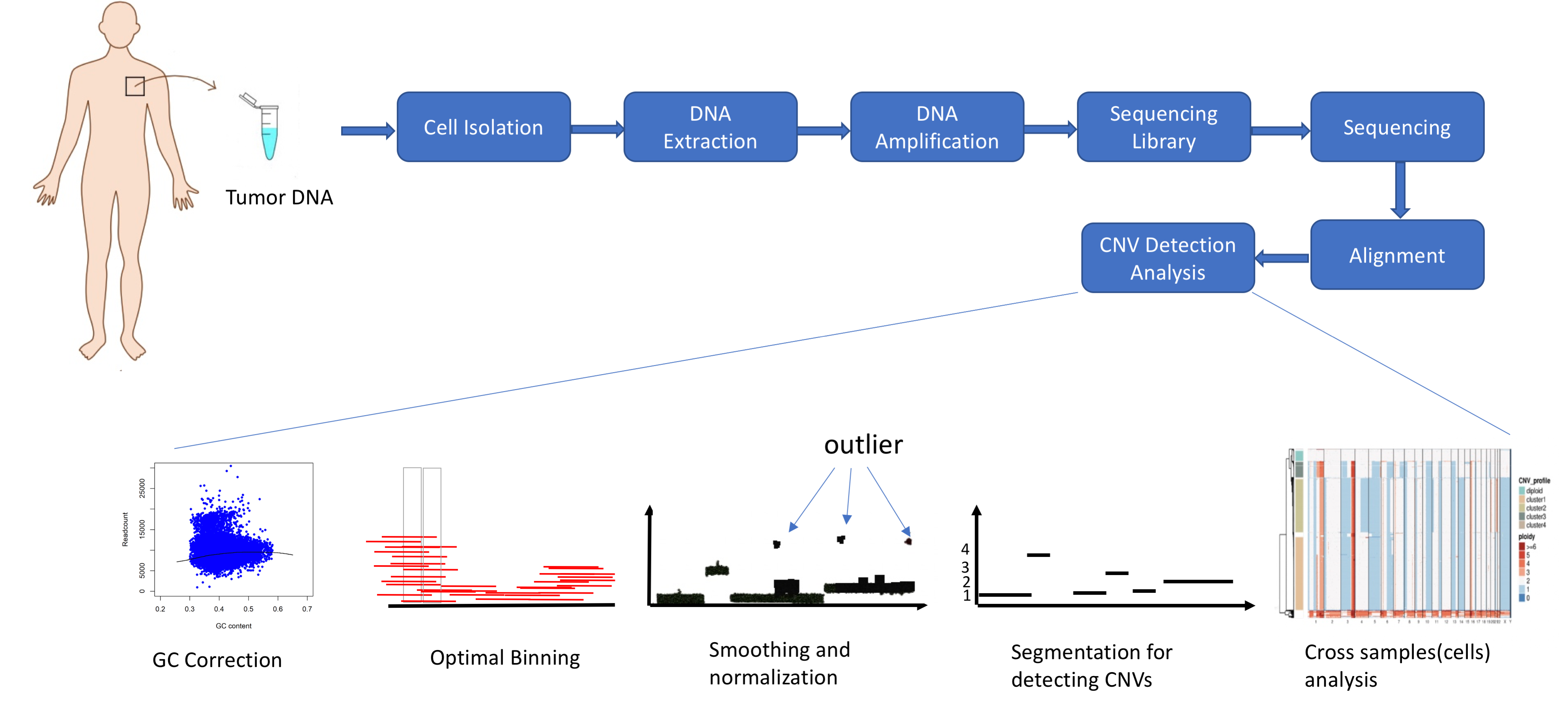
Developing genomic aberration detection methods using DNA single cell sequencing data.
Structural variations (SVs), which include copy number variations (CNVs), translocations and inversions of segments of genome, have gained considerable interest as types of genomic/genetic variation that play an important role in phenotypic diversity, evolution and disease susceptibility in humans and other organisms. With advances in sequencing technologies, Single cell sequencing data are available that have created an opportunity for detecting CNVs and other SVs at the single cell level and thus investigating their roles more thoroughly. However, high levels of noise and biases, low and nonuniform coverage and the "big data" nature of single cell sequencing data present limitations in accurately identifying CNVs using current CNV detection tools. We are working on developing novel CNV detection and visualization methods based on statistical signal processing techniques to identify CNVs more accurately and efficiently.
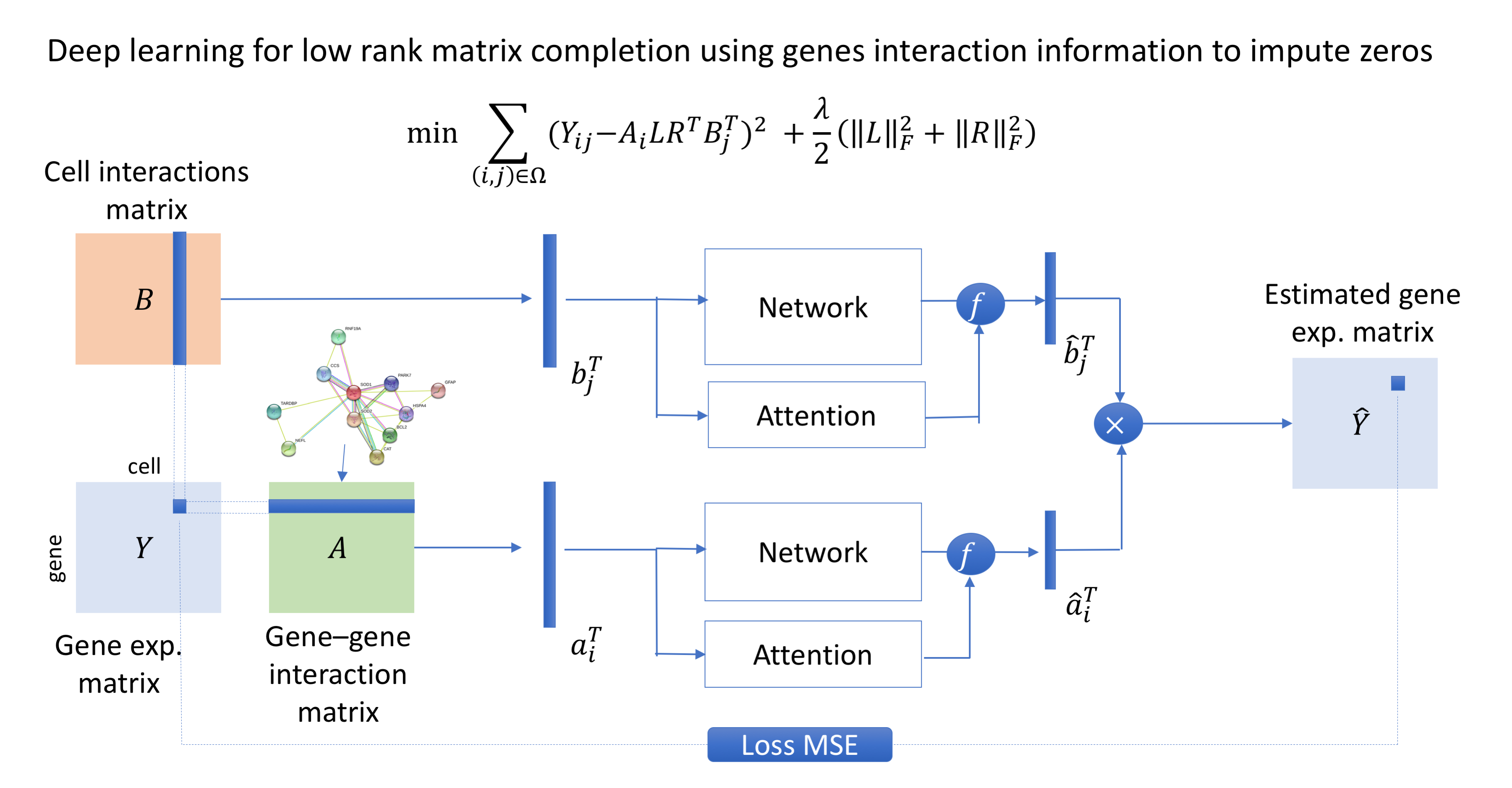
Developing computational methods for analyzing single-cell sequencing data.
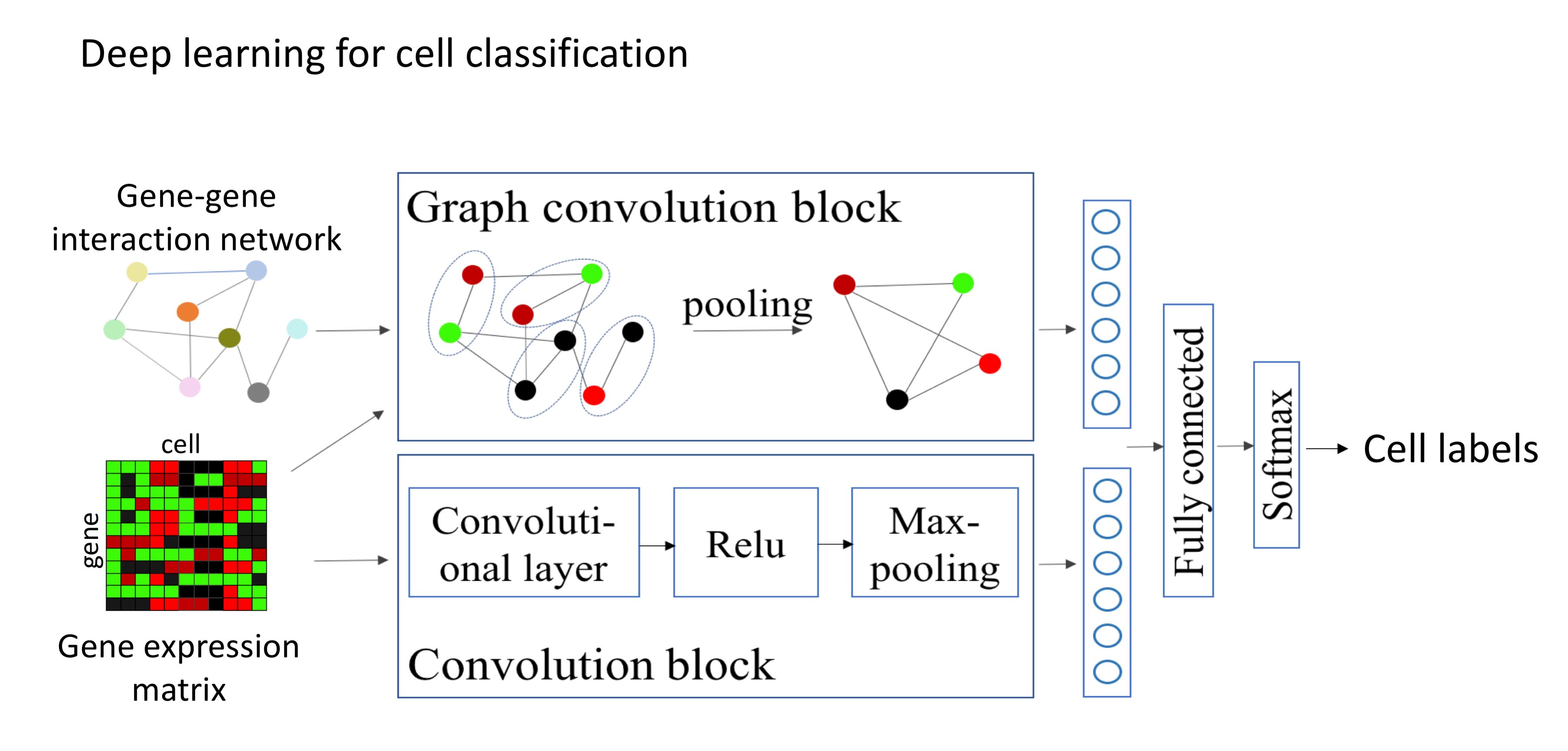
Single-cell sequencing has emerged as a new technique which provides a higher resolution of cellular differences and a better understanding of the function of an individual cell. It applies to several fields such as cancer biology and neurobiology. Even though the structure and format of the data obtained from single-cell sequencing are identical to those from a regular (bulk) sequencing, single-cell sequencing data have introduced new challenges in data analysis, including an abundance of zeros (both biological and technical), increased variability, increased number of samples (thousands to millions of cells) and increased biases. Novel statistical and computational methods are required to address these new challenges. Our current effort is on developing signal processing and machine learning methods for differential gene expression analysis, cell clustering, zero imputation and cell classification.
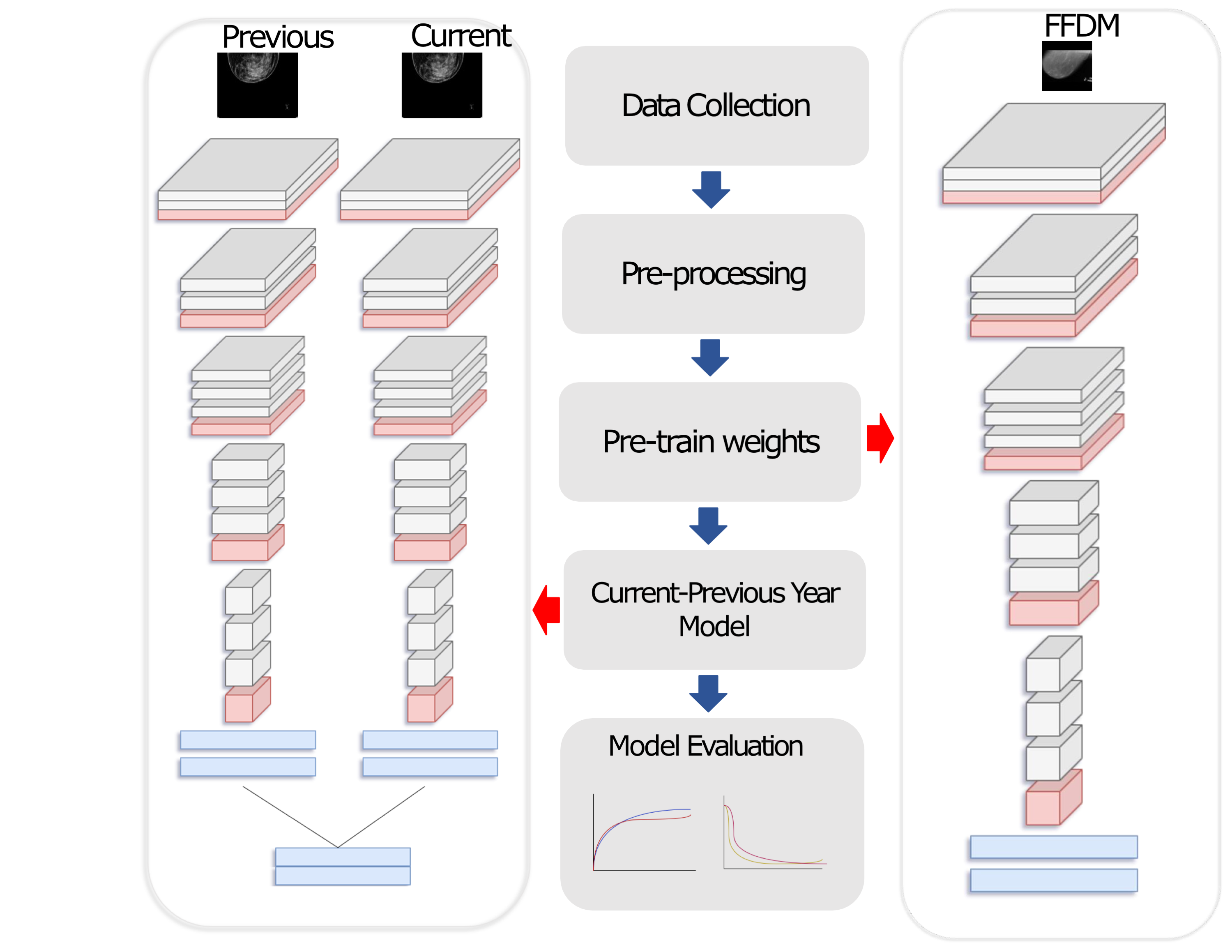
Detection of breast cancer using digital mammograms.
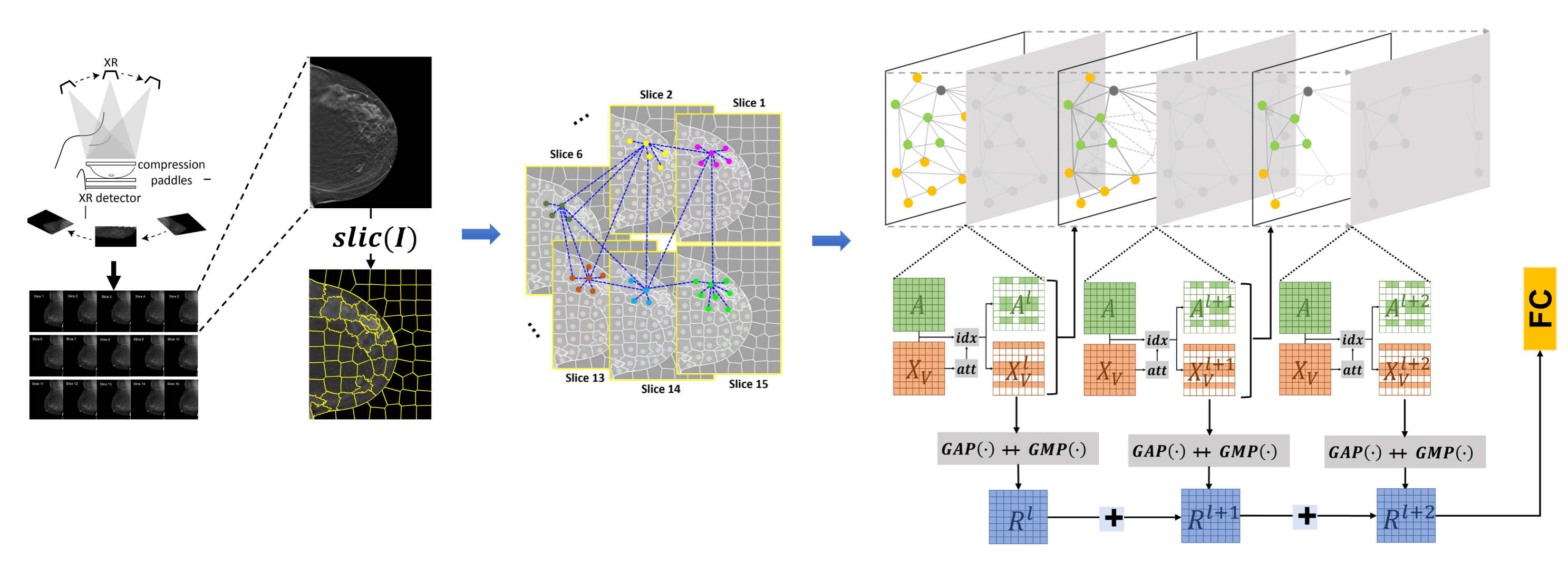
Early diagnosis is a fundamental requirement in order to prevent the increase of deaths caused by breast cancer. Breast cancer screening with mammography has significantly reduced the mortality rates through early detection of the disease. However the complexity of mammograms and the high volume of exams per radiologist have resulted in a significant number of errors. Computer-aided diagnosis (CAD) systems have been very effective to assist radiologist to detect true cancer. However it has been shown that current CAD methods do not improve diagnostic accuracy of mammography. Advances in computational systems, development of new image processing techniques and the availability of digital mammograms open an opportunity for more accurate and more efficient CAD systems. We are working on developing novel CAD methods using deep learning approaches and employing prior mammograms for both 2D mammograms (full-field digital mammography (FFDM)) and 3D mammograms (digital breast tomosynthesis (DBT)).
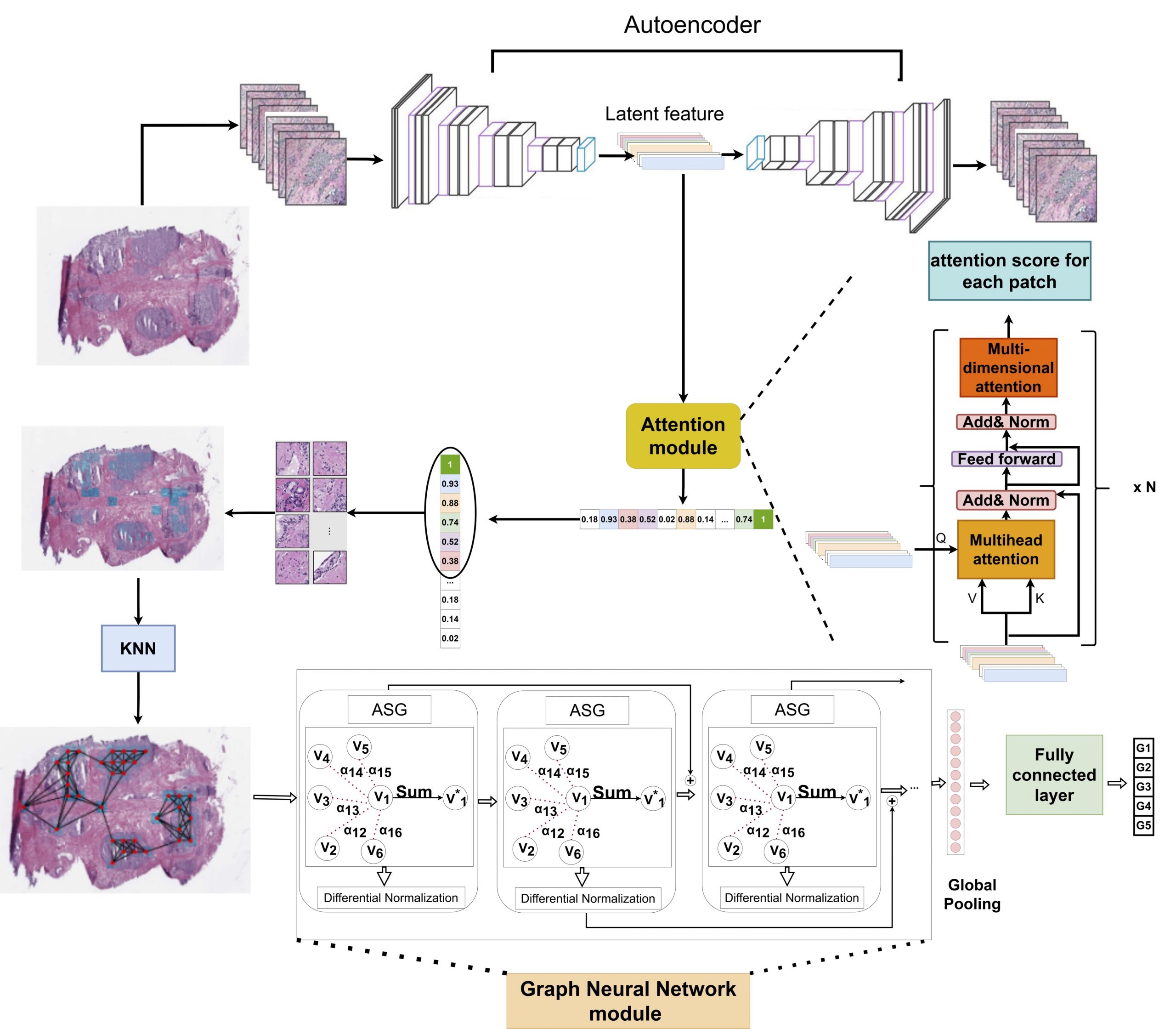
Weakly-Supervised Deep Learning Model for Analyzing Histopathology Images.
Recently deep learning algorithms have been used to analyze histopathology images, delivering promising results. However, most of the algorithms rely on the fully annotated datasets which are expensive to generate. We proposed a novel weakly-supervised algorithm to classify whole slide histology images without having annotated images. The proposed algorithm consists of three steps: (1) extracting discriminative areas in a histopathology image by employing the Multiple Instance Learning (MIL) algorithm based on Transformers, (2) representing the image by constructing a graph using the discriminative patches, and (3) classifying the image into its Gleason grades by developing a Graph Convolutional Neural Network (GCN) based on the gated attention mechanism. We applied the proposed model to the challenging problem of predicting Gleason grades of prostate cancer histology images.
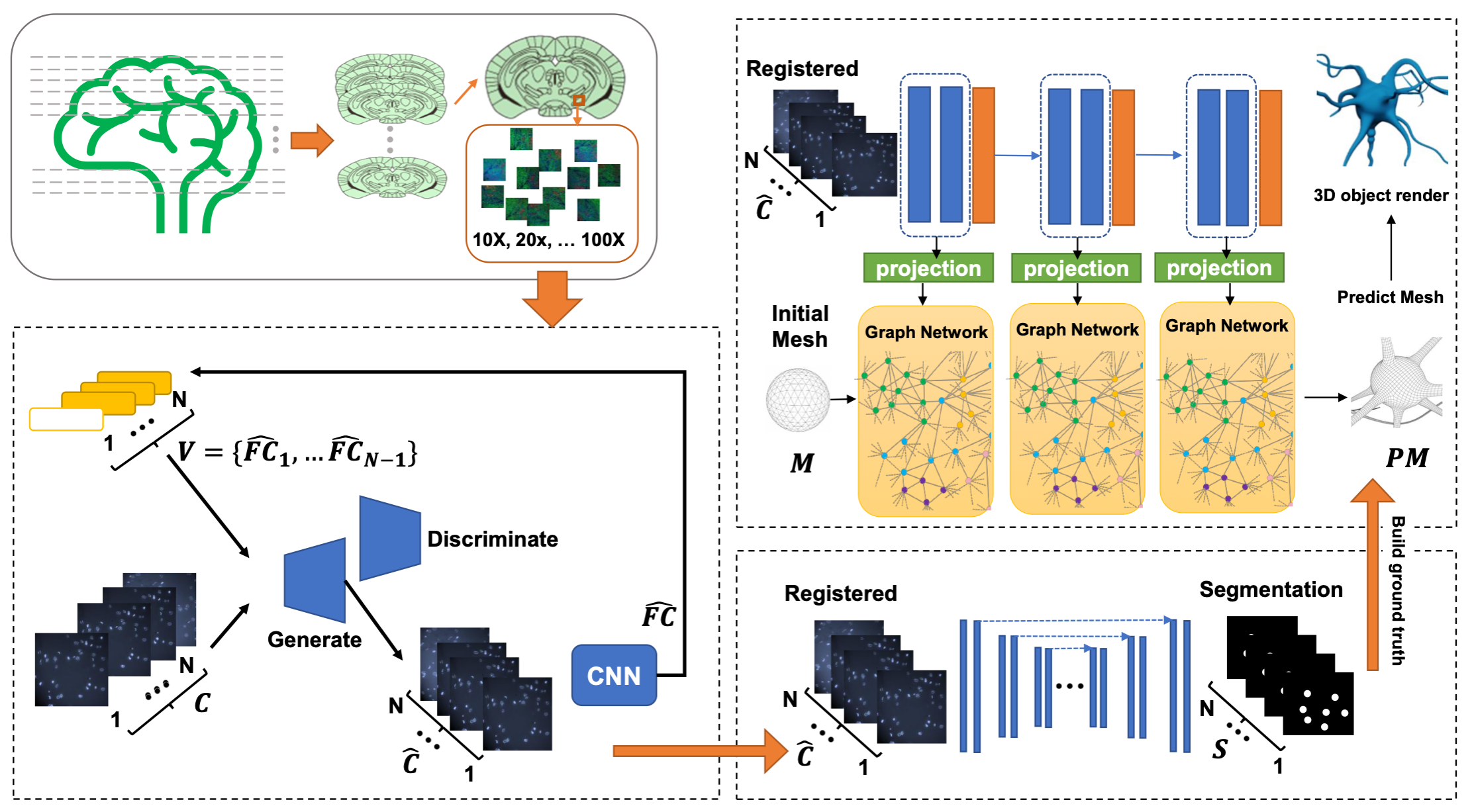
Analyzing mouse brain multiplex microscopic images: Registration, Segmentation, 3D reconstruction